Fenyloketonuria to wrodzona choroba metaboliczna o charakterze dziedzicznym. Podłożem jej jest zaburzenie przemiany fenyloalaniny (aminokwasu egzogennego) w tyrozynę. Wskutek tego obserwuje się nadmierne gromadzenie fenyloalaniny i jej metabolitów we krwi oraz płynach ustrojowych, co powoduje nieodwracalne uszkodzenie ośrodkowego układu nerwowego. Nie ma metody umożliwiających całkowite wyleczenie fenyloketonurii.

Fenyloketonuria – przyczyny
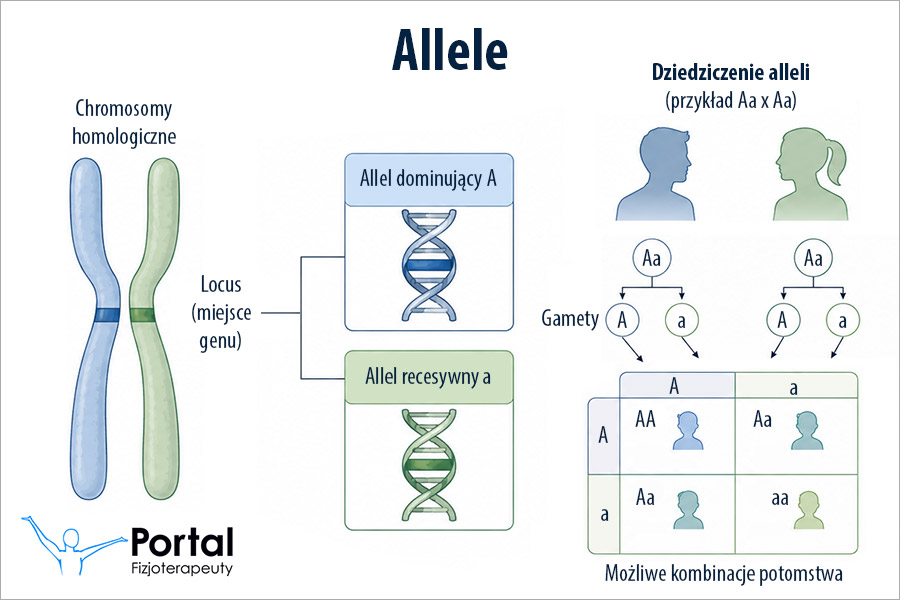
Chorobę dziedziczy się w sposób autosomalny recesywny. Mutacja genetyczna powoduje całkowity lub częściowy brak aktywności hydroksylazy fenyloalaninowej – enzymu wątrobowego katalizującego konwersję aminokwasu fenyloalaniny do tyrozyny. Zaledwie w 3% przypadków mutacja dotyczy enzymów związanych z syntezą lub regeneracją BH4- kofaktora reakcji przekształcania fenyloalaniny w tyrozynę.
Ze względu na wspólny szlak transportowy, nadmiar fenyloalaniny w organizmie prowadzi do zahamowania kompetencyjnego penetracji tyrozyny i tryptofanu, a co się z tym wiąże również metioniny, leucyny, waliny i histydyny przez barierę krew-mózg. Kolejną poważną konsekwencją nadmiaru omawianego aminokwasu jest upośledzenie prawidłowego rozmieszczenia tyrozyny i tryptofanu, co prowadzi do zahamowania transportu przez błony pęcherzyków synaptycznych.
Enzym hydroksylaza fenyloalaniny jest kodowany przez gen PAH, zlokalizowany na 12 chromosomie. Bezpośrednie przyczyny mutacji w przebiegu fenyloketonurii wciąż nie zostały poznane.
Fenyloketonuria – objawy
Ze względu na to, że właściwy poziom fenyloalaniny odkrywa kluczową rolę w rozwoju intelektualnym dziecka, wszelkie zaburzenia w tym zakresie mają poważne konsekwencje. Fenyloketonuria to choroba postępująca i wielonarządowa, dotycząca jednak głównie ośrodkowego układu nerwowego. W okresie noworodkowym i niemowlęcym najczęściej pojawiają się:
- wymioty;
- charakterystyczny „mysi zapach” ciała;
- zmiany skórne o różnym nasileniu, w tym wypryski alergiczne lub łojotokowe.
Wraz z upływem kolejnych miesięcy i lat do obrazu klinicznego dołączają:

- małogłowie;
- drgawki mające postać napadów zgięciowych;
- obniżenie napięcia mięśniowego ze wzmożeniem odruchów;
- nieregularne tiki;
- rytmiczne, wielogodzinne kiwanie się w przód i w tył;
- zaburzenia emocjonalne;
- chód atetotyczny.
Pojawia się również niepełnosprawność intelektualna, bardzo głęboka w przypadku dzieci nieleczonych.
Diagnostyka fenyloketonurii
Podstawą diagnostyki są badania obrazowe mózgu, zwłaszcza rezonans magnetyczny. W jego przebiegu zaobserwować można zmiany w obrębie istoty białej, istoty szarej, wzgórza oraz hipokampa. Od maja 1994 roku wszystkie noworodki urodzone w Polsce objęte są obligatoryjnie 2 testami przesiewowymi w kierunku fenyloketonurii. Wykonuje się je metodą bibułową z krwi pobranej z pięty dziecka. W zależności od uzyskanego wyniku dzieci kwalifikuje się do trzech grup:
- I – stężeniem fenyloalaniny 2-4mg/dL;
- II – stężenie fenyloalaniny 4-8mg/dL, wymagające powtórzenia badania;
- III – stężenie fenyloalaniny powyżej 8mg/dL, takie dzieci wymagają natychmiastowej diagnostyki i leczenia.
Najcięższa postać fenyloketonurii ma miejsce wówczas, gdy stężenie fenyloalaniny przekracza 20 mg/dL. Inną metodą diagnostyczną są badania prenatalne dotyczące oceny genu PAH, wykonywane wówczas, gdy u członka rodziny także stwierdzona została choroba.
Fenyloketonuria – leczenie
Podstawą jest leczenie dietetyczne. Pacjenci tolerują mniej niż 250-350 mg fenyloalaniny zawartej w diecie na dzień, dlatego na ogół konieczne jest korzystanie z pomocy doświadczonego dietetyka klinicznego, który ustali optymalny dla dziecka jadłospis. Przeciwwskazane są m.in.:
- ryby, mięso i jego przetwory;
- produkty zbożowe;
- mleko i inny nabiał;
- orzechy;
- aspartam;
- żelatyna;
- czekolada;
- mak;
- jaja;
- rośliny strączkowe.
Kluczową zasadą jest spożywanie preparatu leczniczego, pokrywającego 70-95% dziennego zapotrzebowania na białko. Żywienie powinno opierać się głównie na tłuszczach. Współcześnie nie ma innych metod leczenia fenyloketonurii. Wspomagająco warto zapisać dziecko do doświadczonego fizjoterapeuty, który będzie prowadzić regularne zajęcia integracji sensorycznej i zadba o prawidłowy rozwój ruchowy.
Polecane produkty:

|
Spirulina + Chlorella – naturalne oczyszczanie organizmu
Spirulina i Chlorella to naturalny produkt, który dostarcza witaminy, minerały, a także inne niezbędne do prawidłowego funkcjonowania składniki odżywcze. Dodatkowo skutecznie wspomaga oczyszczanie organizmu, regulację metabolizmu i wzmacnianie układu … Zobacz więcej... |
Bibliografia
- Jarochowicz S., Mazur A., Fenyloketonuria – choroba metaboliczna uwarunkowana genetycznie, Wydawnictwo UR, Rzeszów 1/2007.
- Kołodziejska A., Czynniki wpływające na jakość i skuteczność leczenia dietetycznego pacjentów z fenyloketonurią, Gdańsk 2018.
- Cabalska B., Wybrane choroby metaboliczne u dzieci, Wydawnictwo Lekarskie PZWL, Warszawa 2002.