Zespół Aperta (ZA), czyli akrocefalosyndaktylia typu pierwszego jest rzadko występującą chorobą genetyczną, a zarazem jedną z najcięższych kraniosynostoz złożonych. Charakterystyczne objawy zespołu opisał w 1906 roku francuski neurolog i pediatra Eugene Apert.
Zespół Aperta – przyczyny
W większości przypadków choroba powstaje w efekcie spontanicznej mutacji w genie. Dziedziczenie jest autosomalne dominujące z równą częstością występowania u chłopców i dziewczynek. Choroba stanowi wynik mutacji w obrębie długiego ramienia chromosomu dziesiątego, w obrębie genu kodującego receptor czynnika wzrostu fibroblastów.
Rzadkość pojawiania się postaci rodzinnej tłumaczy się częstością występowania zaburzeń rozwoju, zmniejszającą prawdopodobieństwo rozmnażania się pacjentów z tym zespołem. Częstość występowania choroby szacuje się na około 15 przypadków na milion żywych urodzeń.
Podsumowując, zespół Aperta pojawia się znikąd, ponieważ w niemal 100% przypadków rodzice są zdrowi i nie wykazują żadnych objawów chorobowych. Mutacja rozwija się zatem samoistnie, bez określonej przyczyny.
Zespół Aperta – objawy
Kliniczne cechy zespołu Aperta można podzielić na kilka klas:
- zaburzenia wzrostu i rozwoju ciała;
- nieprawidłowości w narządach wewnętrznych;
- zaburzenia w obrębie ośrodkowego układu nerwowego;
- zaburzenia rozwojowe i neuropsychologiczne;
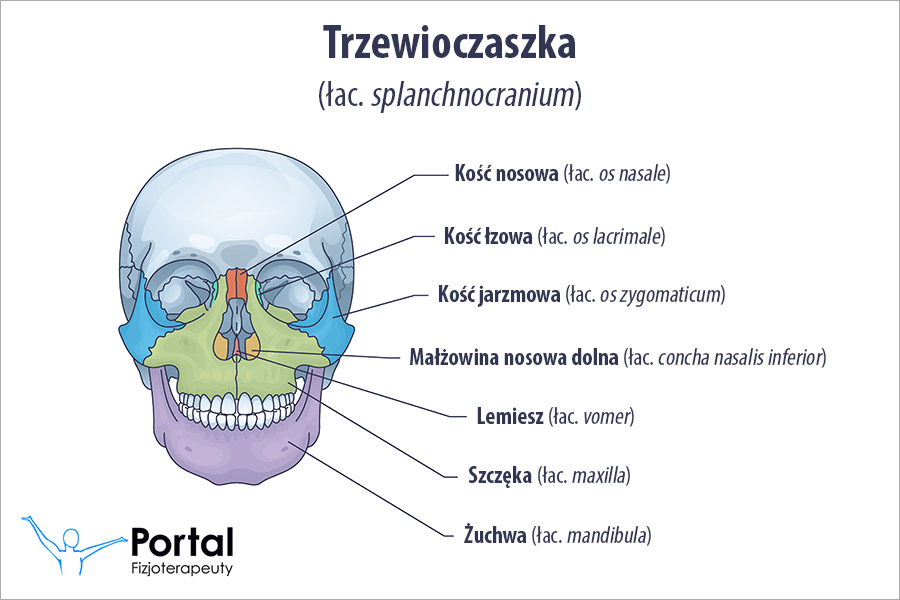
- nieprawidłowości czaszkowo-twarzowe;
- zaburzenia szkieletowe;
- objawy skórne.
Charakterystycznym objawem są zaburzenia wzrostu głowy, w tym nieprawidłowo duży jej obwód oraz deformacja jej wysokości i objętości. Wady sercowo-naczyniowe występują u około 10% dzieci z zespołem. W efekcie złożone i wielokrotne wady serca często wiążą się z wysokim odsetkiem śmiertelności we wczesnym dzieciństwie. Ponadto oczodoły chorych są płaskie i płytkie, co skutkuje wytrzeszczem gałek ocznych.
Inną charakterystyczną cechą zespołu jest hiperteloryzm, czyli powiększenie odstępu między wewnętrznymi kątami szpar powiekowych. Zdarza się, że ze względu na wytrzeszcz, powieki się nie domykają.


Pozostałe, typowe objawy zespołu Aperta to:
- opóźnione wyrzynanie zębów;
- nisko osadzone małżowiny uszne;
- zaburzenia mowy i słuchu;
- obustronne palcozrosty skórne i/lub kostne dłoni i stóp;
- opóźnienie rozwoju intelektualnego;
- zaburzenia oddychania;
- mały nos o zapadniętej nasadzie;
- wysokie i zwężone podniebienie;
- rozszczep podniebienia.
Diagnostyka
Zespół Aperta można diagnozować prenatalnie, ponieważ uwidacznia się w badaniu USG w drugim trymestrze ciąży. U 25-tygodniowego płodu widać pewne cechy mogące wskazywać na chorobę genetyczną, a u 28-tygodniowego płodu zauważa się już zmieniony kształt czaszki i cechy dysmorfii twarzy.
Potwierdzeniem rozpoznania ZA jest badanie cytogenetyczne oraz analiza molekularna genu FGFR2 u płodu na komórkach wyizolowanych z płynu owodniowego lub komórkach z krwi pępowinowej pobranej drogą kordocentezy.
Zespół Aperta – leczenie
Leczenie chorych stanowi spore wyzwanie terapeutyczne, ponadto choroby nie da się wyleczyć. Konieczne jest wielokierunkowe podejście, zindywidualizowane według potrzeb konkretnego pacjenta, ponieważ zauważa się zmienność objawów. Metody lecznicze są zatem dobierane w zależności od występujących objawów.
Przykładowo, przy zaburzeniach oddychania stosuje się wentylację wspomaganą, a w przypadku upośledzonego zamykania oczy zaleca się regularne nawilżanie oczu sztucznymi łzami. Jeśli u pacjentów często występują zakażenia, np. ucha, stosuje się antybiotykoterapię.
Niekiedy wymagane jest leczenie chirurgiczne, np. w przypadku zbyt wczesnego zrośnięcia kości czaszki. Ich operacyjne rozdzielenie redukuje ryzyko nadciśnienia wewnątrzczaszkowego, a także umożliwia prawidłowy rozwój mózgowia.
Jakość życia poprawia odpowiednio dobrana fizjoterapia, przede wszystkim kinezyterapia.
Polecane produkty:

|
Wałek igłowy – aplikator wieloigłowy
Wałek igłowy to aplikator przeznaczony do wykonywania masażu na różne części ciała. Wysoka efektywność znalazła zastosowanie w kompleksowej terapii różnych schorzeń, a także profilaktyce ... Zobacz więcej... |
Bibliografia
- Jakubiuk-Tomaszuk A., Boćkowski L., Sobaniec W., (i inni), Opis przypadku 15-letniej dziewczynki z zespołem Aperta, Neurologia Dziecięca, 21/2012.
- Jakubiuk-Tomaszuk A., Boćkowski L., Sobaniec W., (i inni), Padaczka w zespole Aperta – opis przypadku, Neurologia Dziecięca, 21/2012.
- Hankus A., Larysz D., Zaburzenia neurologopedyczne i neurorozwojowe w dyskraniach syndromicznych na przykładzie zespołu Aperta. Przegląd literatury i opis trzech przypadków, The Clinical Management of Craniosynostosis, 2004.