Zespół Hurler stanowi najcięższą postać mukopolisacharydozy typu 1, choroby lizosomalnej spowodowanej niedoborem enzymów rozkładających glikozaminoglikany. To choroba metaboliczna o podłożu genetycznym, której przyczyny nie zostały poznane. W obrazie klinicznym obserwuje się postępujące pogarszanie stanu zdrowia oraz wiele objawów uniemożliwiających prawidłowe funkcjonowanie. Pacjenci umierają średnio po 10 latach.

Zespół Hurler – przyczyny
Zespół Hurler rozwija się wskutek mutacji w genie IDUA, zlokalizowanym na 4 chromosomie (locus 4p16.3). Mutacja ta powoduje całkowity niedobór enzymu alfa-L-iduronidazy, co z kolei przyczynia się do spichrzania siarczanu heparanu i siarczanu dermatanu w różnych narządach wewnętrznych, w tym w wątrobie czy mięśniu sercowym. Przyczyny i czynniki ryzyka powstawania tej mutacji nie zostały poznane.
Zobacz również: Choroby spichrzeniowe.
Zespół Hurler – objawy
W pierwszych latach życia obserwuje się różnego rodzaju zmiany mięśniowo-szkieletowe, między innymi:
- karłowatość (wzrost organizmu zatrzymuje się całkowicie w okolicach 3. roku życia dziecka);
- objawy dyzostozy;
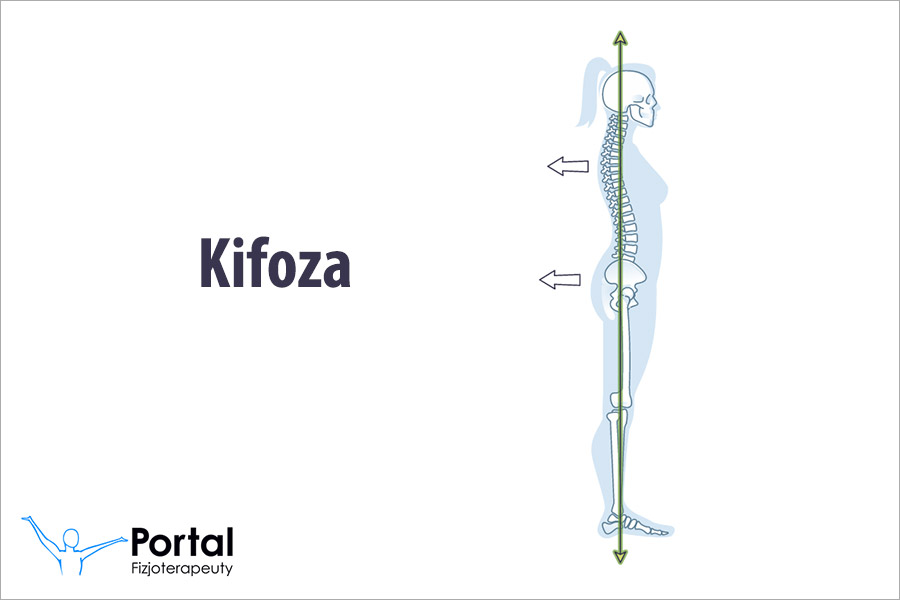
- zwiększoną kifozę piersiowo-krzyżową;
- postępujące pogrubienie rysów twarzy;
- kardiomiopatię;
- nieprawidłowości zastawek serca;
- utratę słuchu;
- powiększeniem migdałków podniebiennych i trzeciego migdałka;
- ciągłą wydzielinę z nosa.
Po ukończeniu 2. roku życia u części pacjentów zauważa się postępujące wodogłowie oraz zmętnienie rogówki wymagające keratoplastyki. Pozostałe objawy, które mogą rozwinąć się w dowolnym wieku, to:


- hirsutyzm;
- przepukliny w różnych częściach ciała;
- powiększenie narządów wewnętrznych;
- zniekształcenie rysów twarzy (płaska twarz, wydatne czoło, obniżona przegroda nosowa);
- nadmiernie powiększony język uniemożliwiający prawidłową wymowę.
U osób chorych pojawia się również upośledzenie umysłowe, często znacznego stopnia. Niedorozwój umysłowy ogranicza kontakt z młodym pacjentem i sprawia, że staje się on całkowicie zależny od rodziców lub opiekunów.
Zespół Hurler – diagnostyka
Bardzo ważna jest diagnostyka różnicowa, która powinna uwzględnić łagodniejszą postać mukopolisacharydozy typu 1, zespół Hurler-Scheie oraz mukopolisacharydozy typu 2 i typu 6. Początkowo rozpoznanie zespołu Hurler może być trudne, ponieważ wiele chorób genetycznych przejawia podobny obraz kliniczny. Pewne rozpoznanie ustala się na podstawie obserwacji zwiększonego wydalania siarczanu heparanu i siarczanu dermatanu w moczu, a także po potwierdzeniu niedoboru enzymu alfa-L-iduronidazy w leukocytach lub fibroblastach. Można wykonać badanie genetyczne, choć nie jest to konieczne.
Zespół Hurler – leczenie
W przypadku dzieci poniżej 2,5 roku życia leczeniem z wyboru jest przeszczep macierzystych komórek krwiotwórczych. Zabieg wydłuża czas przeżycia, pozwala zachować funkcje poznawcze i zmniejsza niektóre objawy somatyczne. Aby zaobserwować oczekiwane efekty przeszczep należy jednak wykonać jak najszybciej, zanim dojdzie do uszkodzenia struktur układu nerwowego. Alternatywnie stosuje się enzymatyczną terapię zastępczą, która jednak nie likwiduje objawów neurologicznych. Bardzo często konieczne są interwencje chirurgiczne (np. przy przepuklinach, problemach zastawek serca czy kompresji rdzenia kręgowego).
Aby dziecko mogło jak najdłużej zachować sprawność oraz aby wspierać jego rozwój w jak największym stopniu, rekomenduje się regularną rehabilitację prowadzoną przez doświadczonego fizjoterapeutę dziecięcego zajmującego się przypadkami neurologicznymi. Ważna jest też dietoterapia i wsparcie psychologiczne dla opiekunów dziecka.
Rokowania zależą od wczesności rozpoznania zespołu. W większości przypadków średni czas przeżycia wynosi 10 lat, choć długość życia można wydłużać za pomocą terapii komórkami macierzystymi. Należy mieć na uwadze, że jest to metoda kosztowna, nierefundowana przez NFZ.
Bibliografia
- Kamyk-Wawryszuk A., Dziecko z mukopolisacharydozą w przedszkolu – perspektywa terapeutów, Studia Edukacyjne, 49/2018.
- Śmigiel R., Szczałuba K., Genetycznie uwarunkowane zaburzenia rozwoju u dzieci, Wydawnictwo Lekarskie PZWL, Warszawa 2021.