Choroba Huntingtona to postępujące schorzenie OUN (ośrodkowego układu nerwowego), dziedziczone autosomalnie dominująco. Charakteryzuje się pląsawiczymi ruchami, postępującym otępieniem i zaburzeniami osobowości. Choroba występuje u 4-8 osób na 100 000.
Choroba Huntingtona – przyczyny
Przyczyną schorzenia są mutacje genu HD kodującego białko zwane huntingtyną. Zmiany chorobowe obejmują różne części ośrodkowego układu nerwowego, najczęściej jednak głównie jądro ogoniaste, skorupę i korę mózgową. Wykazano, że ruchy pląsawicze wiążą się z zaburzeniami czynności układu GABA-ergicznego. To z kolei powoduje zwiększenie liczby powtarzalnych sekwencji trzech nukleotydów CAG. U zdrowego człowieka występuje do 35 kopii CAG, natomiast u chorego na chorobę Huntingtona powtórzeń tych jest 36 lub więcej.
Objawy
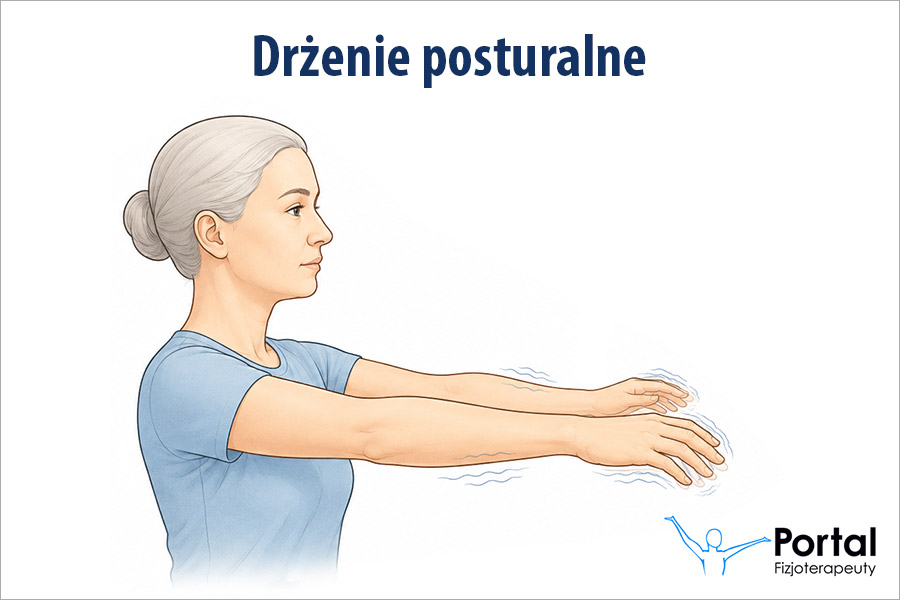
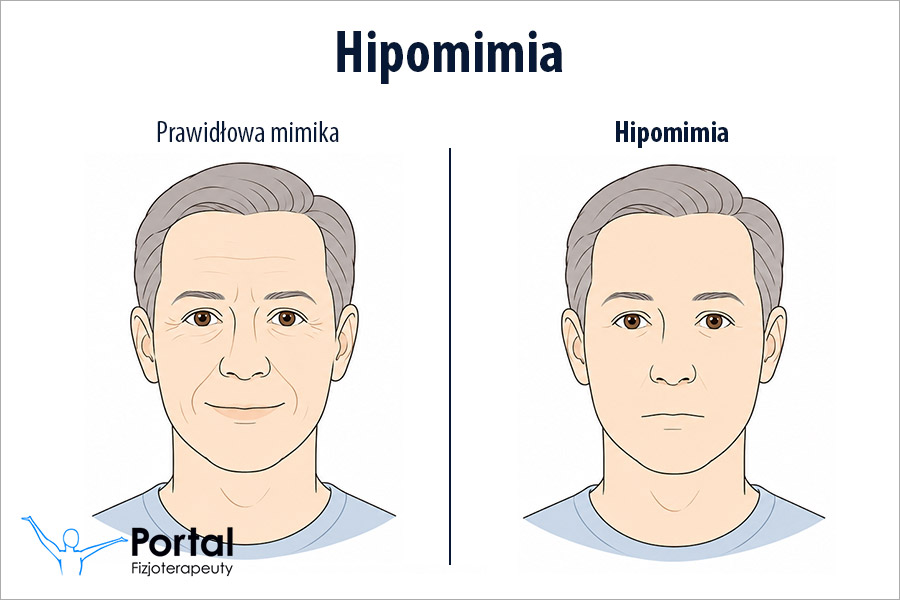
Najbardziej typowym dla tej jednostki chorobowej objawem są ruchy pląsawicze, choć nie występują one u wszystkich pacjentów. Charakterystyczne jest również otępienie, któremu towarzyszą postępujące zaburzenia pamięci. Zauważa się znaczne obniżenie napięcia mięśniowego, co jednak nie występuje w tzw. postaci Westphala, w której stwierdza się bezruch i sztywność. Podsumowując, charakterystyczną dla klasycznej postaci choroby Huntingtona triadą objawów są:
- ruchy pląsawicze;
- zaburzenia funkcji poznawczych;
- zaburzenia psychiczne.
W przebiegu choroby u pacjentów zauważyć można tak zwane zjawisko antycypacji, zwłaszcza przy dziedziczeniu od ojca. Polega ono na coraz wcześniejszym ujawnianiu się choroby w kolejnych pokoleniach. To z kolei wiąże się ze wzrastającą z pokolenia na pokolenie liczbą powtórzeń trinukleotydowych CAG w obrębie genu HD kodującego huntingtynę.
Podstawowe badania laboratoryjne nie wykazują nieprawidłowości. W TK i MRI widać poszerzenie układu komorowego z charakterystycznym obrazem komór bocznych w kształcie skrzydeł motyla. Wynika to z zaniku jąder ogoniastych. Badanie PET (pozytonowa tomografia emisyjna) ujawnia zwolnienie metabolizmu w jądrze ogoniastym, skorupie i korze.
Choroba Huntingtona przebiega powoli, ale postępująco. Przeważają zaburzenia ruchowe, jednak może pojawić się depresja i napady padaczkowe. Z czasem stopniowo dołączają zaburzenia mowy i połykania, a także zaburzenia snu. Chory całkowicie uzależnia się od najbliższego otoczenia, przez co u pacjentów odnotowuje się różnego stopnia zaburzenia depresyjne.

Postacie choroby Huntingtona
W zależności od wieku zachorowania wyróżnia się postać:
- młodzieńczą – zwaną postacią Westphala, przypada na 4-9 rok życia;
- wczesną – przypada na 20-34 rok życia;
- wieku średniego – przypada na 35-49 rok życia;
- późną – dotyczy osób powyżej 50. roku życia.
Inny podział obejmuje objawy kliniczne i wyróżnia postać klasyczną i atypową. W klasycznej występuje charakterystyczna triada objawów, zaś w atypowej dominują bradykinezja, dystonia i objawy zespołu móżdżkowego, przy czym praktycznie nie występują ruchy pląsawicze.
Diagnostyka
Na wstępie należy zróżnicować chorobę z innymi, które mogą objawiać się podobnie. Są to:
- choroba Wilsona;
- wtórne objawowe ruchy pląsawicze;
- choroby autoimmunologiczne, np. pląsawica Sydenhama, toczeń rumieniowaty układowy;
- przewlekłe stosowanie leków, takich jak np. lewodopa;
- zaburzenia metaboliczne i endokrynologiczne;
- choroby naczyniowe, np. udar mózgu w okolicy jądra niskowzgórzowego.
Choroba Huntingtona – leczenie
Leczenie choroby jest wyłącznie objawowe. Przede wszystkim obejmuje rehabilitację. U pacjenta należy zastosować ćwiczenia logopedyczne i kinezyterapię, zwłaszcza reedukację i doskonalenie chodu. Często konieczne jest wsparcie psychologiczne przez całe życie chorego.
Postępowanie farmakologiczne ma na celu głównie osłabienie ruchów pląsawiczych i obejmuje stosowanie neuroleptyków blokujących receptory dopaminergiczne oraz leków usuwających dopaminę z zakończeń presynaptycznych. U niektórych pacjentów konieczne jest wdrożenie farmakoterapii przeciwdepresyjnej i przeciwpsychotycznej, ponieważ tego typu objawy występują u około 10-15% chorych.
Przy odpowiednim leczeniu chorzy przeżywają 15-20 lat od momentu rozpoznania choroby. Przyczyną zgonów są najczęściej powikłania choroby, np. zachłystowe zapalenie płuc.
Polecane produkty:

|
Spirulina + Chlorella – naturalne oczyszczanie organizmu
Spirulina i Chlorella to naturalny produkt, który dostarcza witaminy, minerały, a także inne niezbędne do prawidłowego funkcjonowania składniki odżywcze. Dodatkowo skutecznie wspomaga oczyszczanie organizmu, regulację metabolizmu i wzmacnianie układu … Zobacz więcej... |
Bibliografia
- Adamczak-Ratajczak A., Szumerowicz Ł., Krawczyk M., Mądry E., Choroba Huntingtona – opis przypadku, Family Medicine & Primary Care Review, 2/2011.
- Szczeklik A., Choroby wewnętrzne, Tom II, Wydawnictwo Lekarskie PZWL, Warszawa 2012.
- Kozak-Putowska D., Iłżecka J., Piskorz J., Wójcik G., Problemy zdrowotne chorych na chorobę Huntingtona i ich wpływ na codzienne funkcjonowanie chorego, Medycyna Ogólna i Nauki o Zdrowiu, 2/2016.