Choroba Huntera (zwana także mukopolisacharydozą typu II) jest rzadką chorobą genetyczną objawiającą się wrodzonymi defektami metabolicznymi. To lizosomalna choroba spichrzeniowa związana z chromosomem X.

Choroba Huntera – przyczyny
Omawiana choroba spowodowana jest brakiem lub wadą enzymu sulfatazy iduronianu. Jest on konieczny do rozkładania występujących w organizmie substancji zwanych glikozoaminoglikanami (GAG). Ponieważ pacjenci z zespołem Huntera nie mogą rozkładać tych substancji, GAG stopniowo odkładają się w narządach i uszkadzają je. Powoduje to szereg objawów, w szczególności trudności z oddychaniem i z chodzeniem. Dziedziczenie ma charakter recesywny związany z chromosomem X, w odróżnieniu od innych postaci mukopolisacharydoz, które dziedziczy się autosomalnie recesywnie. Obecnie jest opisanych kilkadziesiąt mutacji znajdujących się na chromosomie X, a nadal wykrywa się nowe, stąd znaczna heterogenność fenotypowa zespołu Huntera.
Choroba dotyczy przede wszystkim mężczyzn, choć odnotowano przypadki zachorowań kobiecych. Szacuje się, że dotyczy 1 na 162 000 żywych urodzeń. Jest schorzeniem przewlekłym i postępującym, co oznacza, że w miarę upływu czasu objawy pogłębiają się, prowadząc do skrócenia życia chorego.
Choroba Huntera – objawy
Objawy choroby Huntera są zróżnicowane i różnią się w poszczególnych przypadkach. To zespół wieloukładowy, może zatem dotyczyć niemal każdego narządu czy tkanki. Do najczęściej występujących objawów klinicznych zalicza się:


- powiększenie języka;
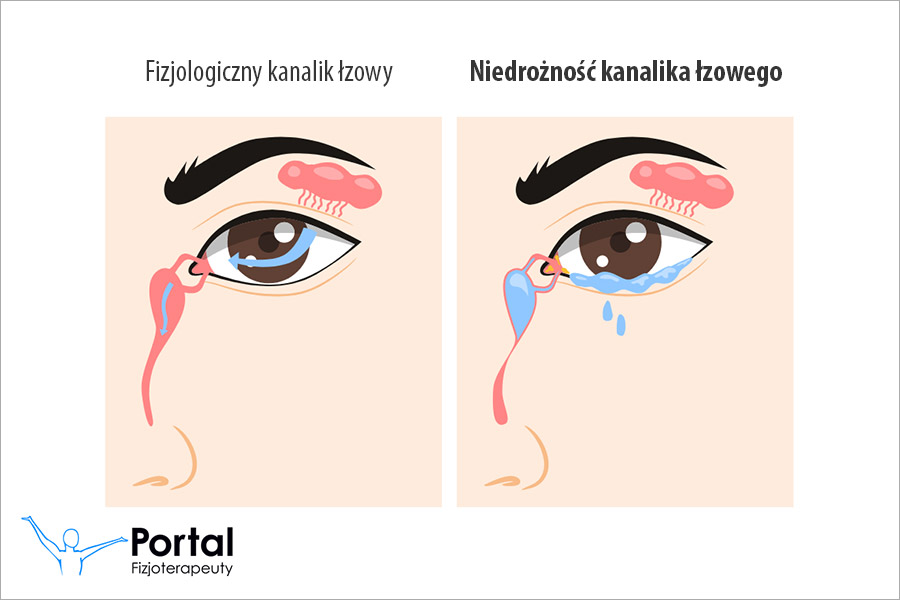
- utratę słuchu;
- poszerzenie rysów twarzy;
- zmniejszenie zakresu ruchomości w stawach;
- powiększenie wątroby lub śledziony;
- wady uzębienia;
- zwężenie górnych dróg oddechowych, czemu może towarzyszyć bezdech senny lub obturacyjna choroba płuc;
- wady serca;
- znaczny niedobór wzrostu;
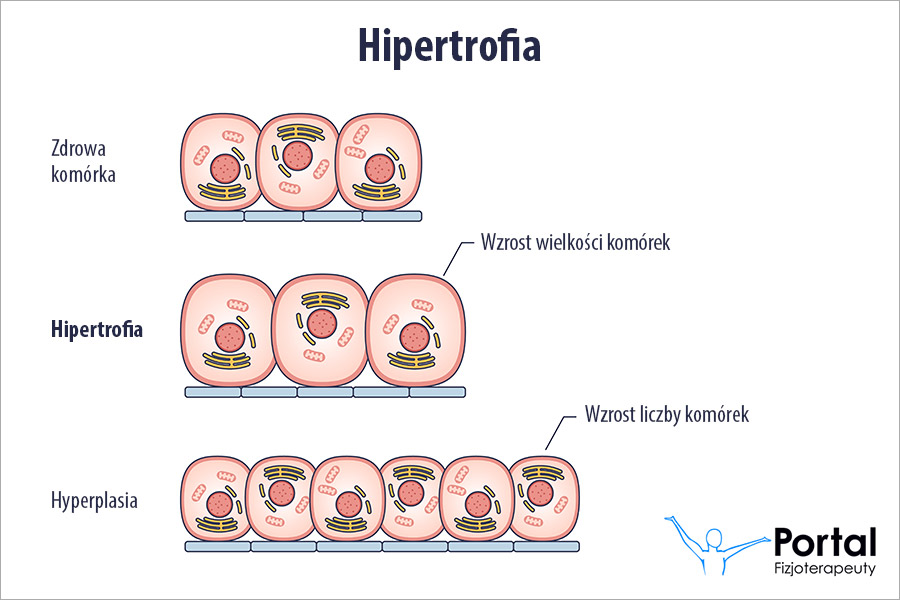
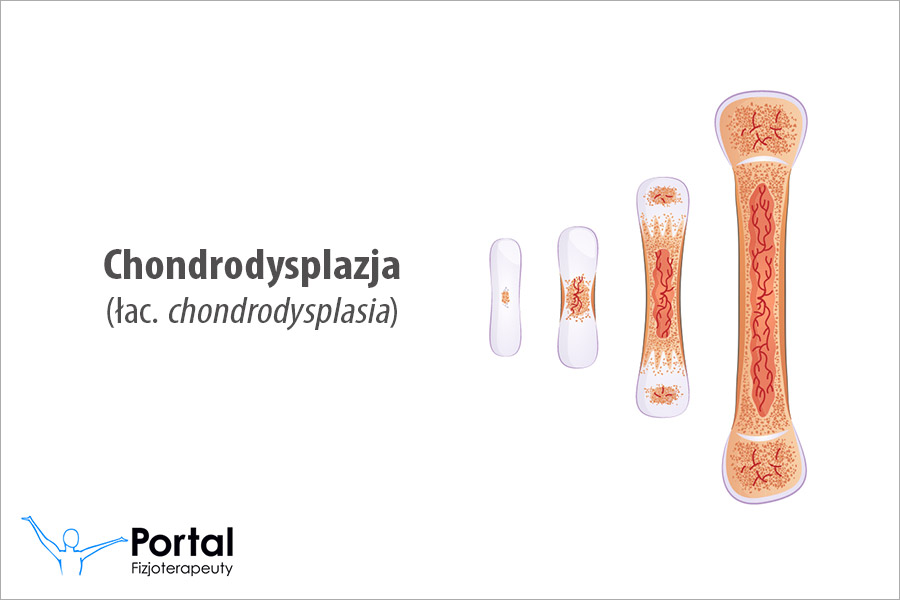
- deformacje w obrębie układu kostnego.
Ze względu na mnogość i powagę objawów choroba znacznie upośledza jakość życia pacjenta. Cierpi on na długotrwałe obniżenie rozwoju fizycznego, poruszanie się sprawia trudności, co pogłębia się wraz z upływem czasu. Ostatecznie chorzy są zmuszeni do korzystania z wózków inwalidzkich. Towarzyszy temu zaburzenie zdolności manualnych – niemożliwe do wykonania stają się czynności względnie proste. Z kolei powiększenie języka utrudnia mowę oraz spożywanie pokarmów. W efekcie, już w młodym wieku chorzy stają się zależni od opiekunów.
W późniejszych stadiach choroby zauważa się objęcie układu nerwowego, co objawia się stopniowym pogarszaniem stanu neurologicznego. Często jest to nasilone współtowarzyszącym wodogłowiem czy zwiększeniem ciśnienia śródczaszkowego.
Choroba Huntera – leczenie
Leczenie choroby Huntera ma charakter objawowy, ponieważ nie jesteśmy w stanie wyleczyć jej przyczyn. Oznacza to, że terapia opiera się najczęściej na fizjoterapii i farmakoterapii – metody te mogą poprawić jakość życia chorego. Fizjoterapia pozwoli na opóźnienie objawów związanych z aparatem ruchu, np. zapewni jak najdłuższe zachowanie ruchomości stawów. Niekiedy wykonuje się zabiegi chirurgiczne, mają one jednak także charakter objawowy. Przykładowo, wykonuje się operację zmniejszającą niedrożność dróg oddechowych.
Choroba prowadzi do śmierci, zazwyczaj w drugiej lub trzeciej dekadzie życia. Przyczyną zgonu jest najczęściej niewydolność serca lub dróg oddechowych.
Polecane produkty:

|
Spirulina + Chlorella – naturalne oczyszczanie organizmu
Spirulina i Chlorella to naturalny produkt, który dostarcza witaminy, minerały, a także inne niezbędne do prawidłowego funkcjonowania składniki odżywcze. Dodatkowo skutecznie wspomaga oczyszczanie organizmu, regulację metabolizmu i wzmacnianie układu … Zobacz więcej... |
Bibliografia
- Pytrus T., Iwańczak B., Wawro J., Czartoryska B., Iwańczak F., Mukopolisacharydoza II (zespół Huntera) – opis przypadku, Adv Clin Exp Med, 2/2006.
- Szczeklik A., Choroby wewnętrzne, Wydawnictwo Medycyna Praktyczna, Kraków 2006.