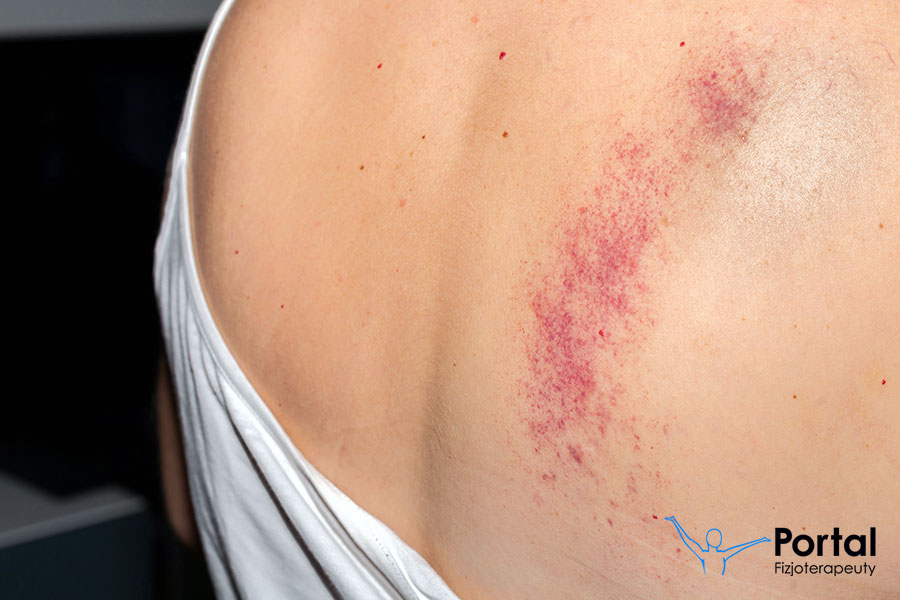
Choroba von Willebranda (zwana wcześniej pseudohemofilią) to najczęściej występująca skaza krwotoczna bez względu na wiek i płeć – występuje 2-krotnie częściej niż hemofilia. Szacuje się, że może dotyczyć nawet do 1,3% populacji. Powoduje nieprawidłowy przebieg procesu krzepnięcia krwi.
Przyczyny
Choroba von Willebranda jest dziedziczona autosomalnie. Przyczyną przedłużonego krwawienia u chorych jest niedobór białka zwanego czynnikiem von Willebranda i oznaczonego w skrócie vWF. Pełni ono funkcję genu. Białko to naturalnie występuje w osoczu krwi osób zdrowych, a także cierpiących na hemofilię.
vWF
Gen vWF lokalizuje się na chromosomie 12, z kolei na chromosomie 22 lokalizuje się pseudogen vWF. Obecność tego pseudogenu znacznie utrudnia analizę patologii choroby. To właśnie wszelkie mutacje białka powodują zmiany strukturalne i odpowiadają za wystąpienie skazy krwotocznej.
Klasyfikacja choroby
Choroba vin Willebranda nie jest jedną, określoną i zawsze tak samą przebiegającą patologią. Dawniej wyróżniano aż 20 rodzajów schorzenia, jednak współcześnie powszechnie przyjęta klasyfikacja obejmuje 7 rodzajów. Są to:
- 1 – częściowy ilościowy niedobór czynnika von Willebranda;
- 2 – defekt jakościowy cząsteczki tego białka;
- 2A – jak wyżej, jednak towarzyszy temu upośledzona adhezja trombocytów zależna od vWF;
- 2B – zwiększone powinowactwo czynnika von Willebranda do glikoproteiny Ib;
- 2M – obniżona adhezja płytek krwi zależna od omawianego czynnika, jednakże bez selektywnego niedoboru wysokocząsteczkowych multimerów vWF;
- 2N – znacznie obniżona zdolność wiązania czynnika VIII;
- 3 – całkowity brak czynnika von Willebranda.
Powyższa klasyfikacja obowiązuje od 2006 roku.


Objawy
Brak lub upośledzenie omawianego białka powoduje znaczne spowolnienie procesu krzepnięcia krwi w porównaniu do osób zdrowych. Cały proces krzepnięcia krwi składa się z kilku etapów, przy czym choroba upośledza jedynie etap 2, 3 i 4. Oznacza to, że naczynia krwionośne osób chorych kurczą się prawidłowo po ich uszkodzeniu (etap pierwszy). Cały problem rozpoczyna się później. W drugim i trzecim etapie krzepnięcia u osoby zdrowej płytki krwi zatrzymują się w miejscu uszkodzenia naczynia, aby rozpocząć formowanie czopu płytkowego. U osoby chorej płytki nie przylegają dość silnie. Ostatni, czwarty etap krzepnięcia polega na tworzeniu trwałego skrzepu, do czego wymagana jest obecność czynnika krzepnięcia VIII. Brak, niedobór lub patologia czynnika von Willebranda upośledza funkcjonowanie czynnika VIII, dlatego utworzenie trwałego skrzepu u chorych trwa znacznie dłużej.
Do alarmujących objawów mogących wskazywać na schorzenie należą:
- przedłużone miesiączkowanie u kobiet;
- łatwe tworzenie siniaków;
- krwawienie z dziąseł czy nosa – z dziąseł np. u dzieci podczas wypadania zębów mlecznych;
- przedłużone krwawienie nawet z niewielkich ran.
Diagnostyka
Osoba podejrzewająca chorobę powinna udać się do hematologa, który wykona wszelkie badania. Należy pamiętać, że zazwyczaj rutynowe badania krwi u osób chorych wychodzą prawidłowo. Badanie powinno być zatem ukierunkowane na ocenę stężenia czynnika von Willebranda i pomiar jego aktywności. Dodatkowo należy sprawdzić aktywność czynnika VIII. Badaniem dodatkowym może być test agregacji płytek krwi. Wymienione badania są czasochłonne i kosztowne. Na wynik niekiedy trzeba czekać nawet kilka tygodni.
Leczenie
Choroba jest nieuleczalna, co oznacza że trwa przez całe życie. Można jednak skutecznie leczyć krwawienia. Wiele osób żyje ze swoją chorobą nieświadomie, aż do momentu większego urazu. W wielu przypadkach choroba nie wpływa na jakość życia, choć wszystko zależy oczywiście od jej rodzaju i nasilenia, co jest kwestią indywidualną. Choroba von Willebranda może mieć postać łagodną lub ciężką.
Leczenie opiera się głównie na farmakoterapii. Dobór leków ściśle zależy od rodzaju choroby. Przykładowo, w typie 1 polecanym lekiem jest desmopresyna, która uwalnia czynnik von Willebranda z wewnętrznej warstwy naczyń krwionośnych. Popularne są również kwasy: aminokapronowy i traneksamowy, które wspomagają utrzymywanie się prawidłowego skrzepu, nie pomagają jednak w samym jego formowaniu jak lek poprzedni. Właśnie dlatego tak ważne jest, aby dobrać odpowiednie środki farmakologiczne indywidualnie do pacjenta i aby pacjent nie stosował ich zamiennie.
Bibliografia
- Balwierz W., Pietrys D., Pawińska-Wąsikowska K., Moryl-Bujakowska A., Choroba von Willebranda u dzieci i młodzieży: diagnostyka i leczenie, Pediatria po Dyplomie, 3/2012.
- Bykowska K., Klasyfikacja i diagnostyka choroby von Willebranda, Hematologia, 1/2013.
Polecane produkty:

|
Spirulina + Chlorella – naturalne oczyszczanie organizmu
Spirulina i Chlorella to naturalny produkt, który dostarcza witaminy, minerały, a także inne niezbędne do prawidłowego funkcjonowania składniki odżywcze. Dodatkowo skutecznie wspomaga oczyszczanie organizmu, regulację metabolizmu i wzmacnianie układu … Zobacz więcej... |

|
Spirulina 100% naturalna
Spirulina platensis - alga o niebiesko-zielonej barwie. Dostarcza kompleks niezwykle ważnych składników odżywczych takich jak m.in. białko, witaminy, minerały, które są niezbędne do prawidłowego funkcjonowania organizmu. Wspomaga regulację metabolizmu … Zobacz więcej... |